Orientation And Equilibrium Disorders
|
Discussion points:
(1) Acute confusion. (2) Notes on encephalitis. (3) Dementia, rapidly progressive dementia, and amnestic syndromes. (4) Notes on prion disease. (5) Equilibrium disorders. (6) Notes on sporadic adult ataxia. (7) The workup of acute confusion, rapidly progressive dementia, and sporadic adult ataxia. (8) Treatment of encephalitis, prion diseases, vertigo, and sporadic adult ataxia. |
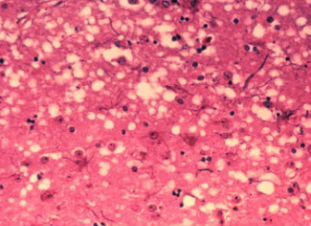
Spongiform "holes" in brain tissue created by prions in Creutzfeldt-Jakob Disease (CJD).
|
Introduction
These often represent a diagnostic challenge. It is important to rise to this challenge so as not to miss a treatable condition.
Acute Confusion
In acute confusion (two types - delirium implies hyperactivity, encephalopathy implies hypoactivity), arousal and attention are depressed to a lesser extent than in coma (use the acronym SEIES). Exclude pain and dehydration first.
(1) Structural/Vascular/Seizures.
- Ischemic stroke, hemorrhage, brain tumour, leptomeningeal metastases, trauma, dementia.
- Hypertensive encephalopathy, PRES, eclampsia, vasculitis, DIC, TTP.
- Postictal, complex partial seizures, non-convulsive status epilepticus.
(2) Electrolytes/Drugs/Alcohol.
- Hyponatremia, hypo/hypercalcemia.
- Sedatives (benzodiazepines, barbiturates, propofol), anticholinergics (TCAs, oxybutinin, benztropine, dimenhydrinate), opiods, beta-blockers, steroids.
- Alcohol intoxication or withdrawal.
(3) Infectious/Autoimmune.
- Infectious meningitis/encephalitis, HIV, VDRL, sepsis.
- Autoimmune encephalitis, sarcoidosis, lupus.
(4) Endocrine/Nutritional.
- Hypo/hyperthyroidism, hypo/hyperglycemia, Wilson's disease, Addison's disease
- Wernicke's encephalopathy, Korsakoff's syndrome, B12/E deficiency.
(5) Syndromes/Organs.
- Serotonin syndrome (antidepressants, opioids), neuroleptic malignant syndrome (antipsychotics, dopamine antagonism/cessation), porphyria.
- Hepatic/uremic/pulmonary encephalopathy.
- Ischemic stroke, hemorrhage, brain tumour, leptomeningeal metastases, trauma, dementia.
- Hypertensive encephalopathy, PRES, eclampsia, vasculitis, DIC, TTP.
- Postictal, complex partial seizures, non-convulsive status epilepticus.
(2) Electrolytes/Drugs/Alcohol.
- Hyponatremia, hypo/hypercalcemia.
- Sedatives (benzodiazepines, barbiturates, propofol), anticholinergics (TCAs, oxybutinin, benztropine, dimenhydrinate), opiods, beta-blockers, steroids.
- Alcohol intoxication or withdrawal.
(3) Infectious/Autoimmune.
- Infectious meningitis/encephalitis, HIV, VDRL, sepsis.
- Autoimmune encephalitis, sarcoidosis, lupus.
(4) Endocrine/Nutritional.
- Hypo/hyperthyroidism, hypo/hyperglycemia, Wilson's disease, Addison's disease
- Wernicke's encephalopathy, Korsakoff's syndrome, B12/E deficiency.
(5) Syndromes/Organs.
- Serotonin syndrome (antidepressants, opioids), neuroleptic malignant syndrome (antipsychotics, dopamine antagonism/cessation), porphyria.
- Hepatic/uremic/pulmonary encephalopathy.
Encephalitis
This is a broad category, especially viral and autoimmune encephalitis.
(1) Viral encephalitis.
- Infection by a virus results in acute inflammation of the brain parenchyma (Solomen et al, 2007). There is a flu-like prodrome followed by symptoms of headache, fever, nausea, vomiting, and disorientation. In more advanced cases, there may be seizures, movement disorders, hallucinations, confusion, and other focal neurological signs. The etiology is usually not identified.
- Viruses are the most common source of encephalitis. In one study with 1570 patients, a confirmed or probable infectious etiology was identified in 16% (69% viral, 20% bacterial, 7% prion, 3% parasitic, and 1% fungal), a possible infectious etiology was identified in 13%, a non-infectious etiology was identified in 8%, and no etiology was identified in 63% (Glaser et al, 2006).
- The most devastating condition is HSV encephalitis from HSV-1 and HSV-2; the latter is only responsible for 5% of cases (Skoldenberg, 1996). These viruses establish a latent infection for the life of the host in the dorsal root ganglia, from which they can reactivate (Steiner, 2011). EEG shows focal abnormalities or diffuse slowing in 80-90% of patients; MRI brain is abnormal in 90% of patients and most commonly shows T2 or FLAIR high signal in the medial temporal lobe; lumbar puncture shows a mononuclear pleocytosis, elevated protein, and positive HSV PCR (sensitivity 94-98%, specificity 98-100%) (Anderson et al, 2014). The HSV PCR results become positive within 24 hours of onset of symptoms and a negative result obtained less than 72 hours after onset of symptoms in a patient with a high pre-test probability should be repeated.
- A milder HSV infection called Mollaret's meningitis results in an aseptic meningitis with attacks of severe headache, meningism, and fever separated by weeks to months; lumbar puncture shows a cerebrospinal fluid pleocytosis (Tyler, 2004).
- An infection called HZV encephalitis from VZV can be an uncommon complication of a herpes zoster shingles infection.
(2) Autoimmune encephalitis.
- A substantial portion of encephalitides are associated with autoantibodies against the extracellular domains of cell-surface proteins critical in the regulation of neuron excitability, including NMDA, VGKC, LGI1, BAGA, AMPA, and CASPR2 (Irani, 2011).
- Patients with limbic encephalitis have detectable antibodies to one of several possible proteins in about half of all cases (Bataller et al, 2006), with paraneoplastic and anti-VGKC antibodies being the most common. Symptoms include subacute short-term memory loss, seizures, confusion, and psychiatric symptoms (Asztely and Kumlien, 2012). Patients with paraneoplastic antibodies have an underlying tumour. Patients with anti-VGKC antibodies are almost always over 50 years of age and have an immunotherapy-responsive syndrome not associated with an underlying tumour (Vincent et al, 2004). A serum hyponatremia may occur. Serum concentrations of anti-VGKC antibodies are far higher than those in the cerebrospinal fluid, which may even be undetectable (anti-VGKC antibodies are also seen in Lambert-Eaton syndrome); MRI brain characteristically shows T2 or FLAIR high signal in the medial temporal lobe. Both hyponatremia and these MRI changes are seen in around 60% of cases (Irani, 2011). Be aware that cancer can produce brainstem or cerebellar encephalitis too, but they do not show the same degree of association with antibodies.
- Patients with anti-NMDA encephalitis are usually young women of African or Asian origin in their twenties or thirties, and over half of these patients have a detectable ovarian teratoma, although a tumour is much rarer in young patients (Titulaer et al, 2013). Patients display a progression of symptoms with (1) a viral-like prodrome of headache and fever, (2) psychiatric symptoms of hallucinations and suicidal ideation, (3) anterograde amnesia, (4) dyskinesias and seizures, (5) catatonia, (6) hypoventilation, and (7) autonomic instability with variatoins in blood pressure and salivation (Gable et al, 2009). EEG is abnormal in most patients and shows nonspecific slow and disorganized activity, sometimes with electrographic seizures; it is not associated with abnormal movements and does not respond to anti-epileptic drugs; MRI brain is normal in 50% of patients, and in the others T2 or FLAIR high signal may be seen in virtually any brain region; lumbar puncture is abnormal in 80% of patients initially and becomes abnormal in nearly all later, with a moderate lymphocytic pleocytosis, normal or mildly increased protein, oligoclonal bands in 60%, and anti-NMDA antibodies in most (Dalmau et al, 2011).
(1) Viral encephalitis.
- Infection by a virus results in acute inflammation of the brain parenchyma (Solomen et al, 2007). There is a flu-like prodrome followed by symptoms of headache, fever, nausea, vomiting, and disorientation. In more advanced cases, there may be seizures, movement disorders, hallucinations, confusion, and other focal neurological signs. The etiology is usually not identified.
- Viruses are the most common source of encephalitis. In one study with 1570 patients, a confirmed or probable infectious etiology was identified in 16% (69% viral, 20% bacterial, 7% prion, 3% parasitic, and 1% fungal), a possible infectious etiology was identified in 13%, a non-infectious etiology was identified in 8%, and no etiology was identified in 63% (Glaser et al, 2006).
- The most devastating condition is HSV encephalitis from HSV-1 and HSV-2; the latter is only responsible for 5% of cases (Skoldenberg, 1996). These viruses establish a latent infection for the life of the host in the dorsal root ganglia, from which they can reactivate (Steiner, 2011). EEG shows focal abnormalities or diffuse slowing in 80-90% of patients; MRI brain is abnormal in 90% of patients and most commonly shows T2 or FLAIR high signal in the medial temporal lobe; lumbar puncture shows a mononuclear pleocytosis, elevated protein, and positive HSV PCR (sensitivity 94-98%, specificity 98-100%) (Anderson et al, 2014). The HSV PCR results become positive within 24 hours of onset of symptoms and a negative result obtained less than 72 hours after onset of symptoms in a patient with a high pre-test probability should be repeated.
- A milder HSV infection called Mollaret's meningitis results in an aseptic meningitis with attacks of severe headache, meningism, and fever separated by weeks to months; lumbar puncture shows a cerebrospinal fluid pleocytosis (Tyler, 2004).
- An infection called HZV encephalitis from VZV can be an uncommon complication of a herpes zoster shingles infection.
(2) Autoimmune encephalitis.
- A substantial portion of encephalitides are associated with autoantibodies against the extracellular domains of cell-surface proteins critical in the regulation of neuron excitability, including NMDA, VGKC, LGI1, BAGA, AMPA, and CASPR2 (Irani, 2011).
- Patients with limbic encephalitis have detectable antibodies to one of several possible proteins in about half of all cases (Bataller et al, 2006), with paraneoplastic and anti-VGKC antibodies being the most common. Symptoms include subacute short-term memory loss, seizures, confusion, and psychiatric symptoms (Asztely and Kumlien, 2012). Patients with paraneoplastic antibodies have an underlying tumour. Patients with anti-VGKC antibodies are almost always over 50 years of age and have an immunotherapy-responsive syndrome not associated with an underlying tumour (Vincent et al, 2004). A serum hyponatremia may occur. Serum concentrations of anti-VGKC antibodies are far higher than those in the cerebrospinal fluid, which may even be undetectable (anti-VGKC antibodies are also seen in Lambert-Eaton syndrome); MRI brain characteristically shows T2 or FLAIR high signal in the medial temporal lobe. Both hyponatremia and these MRI changes are seen in around 60% of cases (Irani, 2011). Be aware that cancer can produce brainstem or cerebellar encephalitis too, but they do not show the same degree of association with antibodies.
- Patients with anti-NMDA encephalitis are usually young women of African or Asian origin in their twenties or thirties, and over half of these patients have a detectable ovarian teratoma, although a tumour is much rarer in young patients (Titulaer et al, 2013). Patients display a progression of symptoms with (1) a viral-like prodrome of headache and fever, (2) psychiatric symptoms of hallucinations and suicidal ideation, (3) anterograde amnesia, (4) dyskinesias and seizures, (5) catatonia, (6) hypoventilation, and (7) autonomic instability with variatoins in blood pressure and salivation (Gable et al, 2009). EEG is abnormal in most patients and shows nonspecific slow and disorganized activity, sometimes with electrographic seizures; it is not associated with abnormal movements and does not respond to anti-epileptic drugs; MRI brain is normal in 50% of patients, and in the others T2 or FLAIR high signal may be seen in virtually any brain region; lumbar puncture is abnormal in 80% of patients initially and becomes abnormal in nearly all later, with a moderate lymphocytic pleocytosis, normal or mildly increased protein, oligoclonal bands in 60%, and anti-NMDA antibodies in most (Dalmau et al, 2011).
Dementia
In dementia (progressive cognitive impairment) arousal and attention are usually preserved.
(1) Classic dementia syndromes.
- Alzheimer's dementia.
- Vascular dementia (strokes within the thalamus/hippocampus/neocortex, Binswanger disease, cerebral amyloid angiopathy).
- Frontotemporal dementia.
(2) Movement disorders.
- Parkinson's disease with dementia, dementia with Lewy bodies.
- PSP, MSA, CBD.
- Huntington's disease.
- Alzheimer's dementia.
- Vascular dementia (strokes within the thalamus/hippocampus/neocortex, Binswanger disease, cerebral amyloid angiopathy).
- Frontotemporal dementia.
(2) Movement disorders.
- Parkinson's disease with dementia, dementia with Lewy bodies.
- PSP, MSA, CBD.
- Huntington's disease.
In a rapidly progressive dementia (rapid progressive cognitive impairment) the etiologies for acute confusion are relevant (use the acronym SEIES). In addition, consider normal pressure hydrocephalus, dialysis dementia, CJD, and depression as they could escape unnoticed.
In amnestic syndromes (disorders of memory) there may be a coexisting delirium/dementia, or the memory loss may be the only presenting feature. The differentials are similar to those described above.
In amnestic syndromes (disorders of memory) there may be a coexisting delirium/dementia, or the memory loss may be the only presenting feature. The differentials are similar to those described above.
Prion Disease
Prions are misfolded proteins that replicate by converting the properly folded proteins in their host to the same misfolded structure they possess. The most common prion disease in humans is Creutzfeldt-Jakob disease (CJD), a degenerative neurological disorder that results from the prions triggering neuron degeneration such that the brain tissue develops holes, taking on a sponge-like architecture.
(1) Sporadic CJD.
- The most common (85% of CJD patients) and presents around 60 years of age with a rapid progressive dementia. EEG shows nonspecific findings such as focal followed by diffuse slowing early on, subsequently periodic sharp waves usually at 1 Hz that eventually dominate and may be associated with myoclonus, and lastly the decline and disappearance of the discharges leaving a featureless record (Geschwind et al, 2008; Weiser et al, 2006); MRI brain shows hyperintensities in the caudate nucleus and putamen (91% sensitive, 94% specific); lumbar puncture is over 95% positive for 14-3-3 tau protein; and tonsillar biopsy is negative. Median illness duration is 4-5 months (Corato et al, 2006; Geschwind et al, 2008).
(2) Familial CJD.
- The next most common (10-15%) and is an inherited form that presents in a similar fashion to sporadic CJD. The age of onset is generally younger and the course of illness longer.
(3) Variant CJD.
- Presents with profound psychiatric illness, followed by a movement disorder and/or ataxia (Geschwind et al, 2008). EEG only rarely shows PSWCs; MRI brain shows hyperintensity in the thalamic pulvinar nuclei; lumbar puncture is only 50% positive for 14-3-3 tau protein; and tonsillar biopsy is positive (diagnosis is therefore by tonsillar biopsy). Median illness duration is 13 months (Corato et al, 2006).
(4) Iatrogenic CJD.
- Arises from contamination with tissue from an infected person, usually from a medical procedure such as hormone use or transplants, and it presents in a similar fashion to sporadic CJD.
(1) Sporadic CJD.
- The most common (85% of CJD patients) and presents around 60 years of age with a rapid progressive dementia. EEG shows nonspecific findings such as focal followed by diffuse slowing early on, subsequently periodic sharp waves usually at 1 Hz that eventually dominate and may be associated with myoclonus, and lastly the decline and disappearance of the discharges leaving a featureless record (Geschwind et al, 2008; Weiser et al, 2006); MRI brain shows hyperintensities in the caudate nucleus and putamen (91% sensitive, 94% specific); lumbar puncture is over 95% positive for 14-3-3 tau protein; and tonsillar biopsy is negative. Median illness duration is 4-5 months (Corato et al, 2006; Geschwind et al, 2008).
(2) Familial CJD.
- The next most common (10-15%) and is an inherited form that presents in a similar fashion to sporadic CJD. The age of onset is generally younger and the course of illness longer.
(3) Variant CJD.
- Presents with profound psychiatric illness, followed by a movement disorder and/or ataxia (Geschwind et al, 2008). EEG only rarely shows PSWCs; MRI brain shows hyperintensity in the thalamic pulvinar nuclei; lumbar puncture is only 50% positive for 14-3-3 tau protein; and tonsillar biopsy is positive (diagnosis is therefore by tonsillar biopsy). Median illness duration is 13 months (Corato et al, 2006).
(4) Iatrogenic CJD.
- Arises from contamination with tissue from an infected person, usually from a medical procedure such as hormone use or transplants, and it presents in a similar fashion to sporadic CJD.
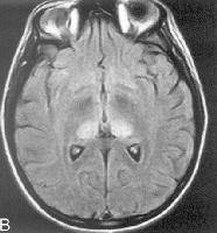
|
The pulvinar sign - this MRI brain shows bilateral hyperintensities involving the thalamic pulvinar nuclei bilaterally, classically seen in variant CJD.
|
Equilibrium
Equilibrium is the ability to maintain body orientation in space and it relies upon continuous visual, vestibular (inner ear labyrinth and vestibular nerve), and proprioceptive (sensory nerves, sensory nerve roots, posterior columns of the spinal cord, and medial lemniscus of the medulla - and even the thalamus and parietal lobe) input all of which is integrated in the brainstem and cerebellum. Equilibrium disorders result from damage to one or more of these structures resulting in vertigo and/or ataxia. When a patient says they are "dizzy," clarify if they have presyncope, vertigo, or imbalance (usually from ataxia).
(1) Vertigo.
- Vertigo is an illusory sense of movement of the body or the environment. It is often associated with nystagmus (rhythmic oscillation of the eyes). It should be localized to the peripheral (vestibular) or central pathways (brainstem or cerebellum).
- Peripheral vertigo is often intermittent, severe, and associated with horizontal unidirectional nystagmus, hearing loss, and tinnitus.
- Central vertigo is often constant, mild to moderate, and associated with horizontal/vertical uni/bidirectional nystagmus and brainstem or cerebellar signs.
- The differentials (in order of prevalence) are benign positional vertigo (lasts <1 minute), functional (think they can partially control it, not severe), central, vestibular migraine (lasts 5 minutes to 72 hours), most have at least one of headache, photophobia, phonopobia, visual aura), Meniere's (lasts 20 minutes to 12 hours, sensorineural hearing loss, fullness), vestibulopathy (unilateral or bilateral, lasts hours to days, visual tilting, worse when moving, head thrust positive), and vestibular paroxysmia (numerous attacks lasting seconds to minutes).
(2) Ataxia.
- Ataxia is incoordination of movement that is not the result of muscle weakness. It should be classified as vestibular, cerebellar, or sensory/proprioceptive.
- Vestibular ataxia displays vertigo and nystagmus, and the patient may have a borderline Romberg's.
- Cerebellar ataxia may display vertigo and nystagmus, there is often dysarthria or limb ataxia, and the patient will be unable to stand even with eyes open.
- Sensory ataxia displays numbness/tingling and limb ataxia particularly in the legs, impaired position sense and ankle reflexes, and a positive Romberg's.
It is useful to know the cerebellar exam.
(1) Vertigo.
- Vertigo is an illusory sense of movement of the body or the environment. It is often associated with nystagmus (rhythmic oscillation of the eyes). It should be localized to the peripheral (vestibular) or central pathways (brainstem or cerebellum).
- Peripheral vertigo is often intermittent, severe, and associated with horizontal unidirectional nystagmus, hearing loss, and tinnitus.
- Central vertigo is often constant, mild to moderate, and associated with horizontal/vertical uni/bidirectional nystagmus and brainstem or cerebellar signs.
- The differentials (in order of prevalence) are benign positional vertigo (lasts <1 minute), functional (think they can partially control it, not severe), central, vestibular migraine (lasts 5 minutes to 72 hours), most have at least one of headache, photophobia, phonopobia, visual aura), Meniere's (lasts 20 minutes to 12 hours, sensorineural hearing loss, fullness), vestibulopathy (unilateral or bilateral, lasts hours to days, visual tilting, worse when moving, head thrust positive), and vestibular paroxysmia (numerous attacks lasting seconds to minutes).
(2) Ataxia.
- Ataxia is incoordination of movement that is not the result of muscle weakness. It should be classified as vestibular, cerebellar, or sensory/proprioceptive.
- Vestibular ataxia displays vertigo and nystagmus, and the patient may have a borderline Romberg's.
- Cerebellar ataxia may display vertigo and nystagmus, there is often dysarthria or limb ataxia, and the patient will be unable to stand even with eyes open.
- Sensory ataxia displays numbness/tingling and limb ataxia particularly in the legs, impaired position sense and ankle reflexes, and a positive Romberg's.
It is useful to know the cerebellar exam.
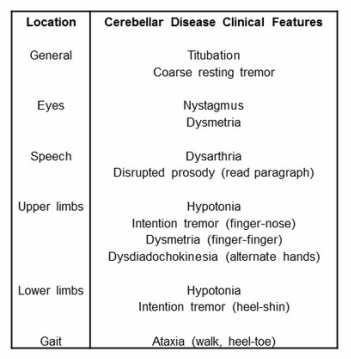
Equilibrium disorders may be grouped according to peripheral/vestibular vertigo or ataxia, central/cerebellar vertigoor ataxia, and sensory ataxia.
(1) Peripheral/vestibular vertigo or ataxia.
- Benign positional vertigo (episodic attacks and related to position, no hearing loss).
- Meniere's disease (episodic attacks with insidious fullness, tinnitus, and sensorineural hearing loss).
- Acute peripheral vestibulopathy (spontaneous attack up to two weeks that resolves, no hearing loss).
- Toxic vestibulopathies (alcohol, aminoglycosides, salicylates, quinine, cisplatin).
- Cerebellopontine angle tumour (insidious unilateral sensorineural hearing loss).
- Otosclerosis (similar to Meniere's, may have conductive or sensorineural hearing loss).
- Head trauma (may have conductive or sensorineural hearing loss).
(2) Acute central/cerebellar vertigo or ataxia.
- Drug intoxication (alcohol, benzodiazepines,and AEDs).
- Stroke (internal auditory occlusion, midbrain, lateral medulla, cerebellum) or cerebellar hemorrhage.
- Inflammation (viral, bacterial, ADEM, and Miller Fisher syndrome).
- Wernicke's encephalopathy.
(3) Chronic central/cerebellar vertigo or ataxia.
- Vestibular migraine (episodic vertigo with migraine).
- Structural (multiple sclerosis, posterior fossa tumour, vascular malformation).
- Sporadic adult ataxia (alcohol/toxin/paraneoplastic, B12/E deficiency, superficial siderosis, chronic infection, SCAs, Friedreich's, FXTAS, mitochondrial, MSA, SAOA).
- Hypothyroidism, Wilson's disease.
(4) Sensory ataxia.
- Sensory neuropathy or neuronopathy (paraneoplastic, isoniazid, taxol, B6 toxicity, Miller Fisher syndrome, anti-MAG).
- Myelopathy (multiple sclerosis, transverse myelitis, cord compression from tumour or vascular malformation).
- Combined peripheral/central lesions (Friedreich's, tabes dorsalis, nitrous oxide, B12/E deficiency).
- Friedreich's ataxia (prominent sensory impairment, areflexia, kyphoscoliosis, and cardiomyopathy).
- Benign positional vertigo (episodic attacks and related to position, no hearing loss).
- Meniere's disease (episodic attacks with insidious fullness, tinnitus, and sensorineural hearing loss).
- Acute peripheral vestibulopathy (spontaneous attack up to two weeks that resolves, no hearing loss).
- Toxic vestibulopathies (alcohol, aminoglycosides, salicylates, quinine, cisplatin).
- Cerebellopontine angle tumour (insidious unilateral sensorineural hearing loss).
- Otosclerosis (similar to Meniere's, may have conductive or sensorineural hearing loss).
- Head trauma (may have conductive or sensorineural hearing loss).
(2) Acute central/cerebellar vertigo or ataxia.
- Drug intoxication (alcohol, benzodiazepines,and AEDs).
- Stroke (internal auditory occlusion, midbrain, lateral medulla, cerebellum) or cerebellar hemorrhage.
- Inflammation (viral, bacterial, ADEM, and Miller Fisher syndrome).
- Wernicke's encephalopathy.
(3) Chronic central/cerebellar vertigo or ataxia.
- Vestibular migraine (episodic vertigo with migraine).
- Structural (multiple sclerosis, posterior fossa tumour, vascular malformation).
- Sporadic adult ataxia (alcohol/toxin/paraneoplastic, B12/E deficiency, superficial siderosis, chronic infection, SCAs, Friedreich's, FXTAS, mitochondrial, MSA, SAOA).
- Hypothyroidism, Wilson's disease.
(4) Sensory ataxia.
- Sensory neuropathy or neuronopathy (paraneoplastic, isoniazid, taxol, B6 toxicity, Miller Fisher syndrome, anti-MAG).
- Myelopathy (multiple sclerosis, transverse myelitis, cord compression from tumour or vascular malformation).
- Combined peripheral/central lesions (Friedreich's, tabes dorsalis, nitrous oxide, B12/E deficiency).
- Friedreich's ataxia (prominent sensory impairment, areflexia, kyphoscoliosis, and cardiomyopathy).
Sporadic Adult Ataxia
The sporadic adult ataxias are a heterogenous group of 50 to 100 disorders in which progressive ataxia is prominent; they usually present after 40 years of age (Klockgether, 2010). Diagnosis can be a clinical challenge.
(1) Acquired.
- Alcoholic cerebellar degeneration (including B1 deficiency) occurs in 11-27% of alcoholics and presents with severe gait and lower limb ataxia, with relatively little effect on ocular eye movement, speech, or upper limbs; MRI shows vermal cerebellar atrophy (Klockgether, 2010).
- Toxins such as phenytoin, amiodarone, chemotherapy, lithium, and heavy metals are also possible etiologies.
- Paraneoplastic cerebellar degeneration may result from almost any tumour or hematological malignancy and presents with a subacute ataxia over several weeks to months. Further immune-related etiologies include anti-GAD ataxia (associated with diabetes), gluten ataxia (associated with celiac disease), and autoimmune thyroiditis or SREAT.
- Vitamin B12 and E deficiencies often present with a neuropathy as well.
- Superficial siderosis from repeated subarachnoid bleeds in the setting of tumours or vascular abnormalitie; MRI brain shows a linear hypointensity on the surface of the brain or spinal cord (Klockgether, 2010).
- Chronic infections such as neurosyphilis, Whipple's, HIV, and CJD are possible etiologies.
(2) Hereditary.
- Friedreich's ataxia results in a sensory ataxia; MRI shows atrophy of the cervical spine.
- Spinocerebellar ataxias (SCAs) are autosomal dominant ataxias produce a cerebellar syndrome and are often associated with additional findings.
- Fragile X-associated tremor/ataxia syndrome (FXTAS) produces an ataxia with a prominent tremor, often with cognitive decline, parkinsonism, neuropathy, and autonomic failure.
- Mitochondrial disorders such as MELAS, MERFF, and NARP are possible etiologies.
(3) Sporadic degenerative.
- Multiple systems atrophy (MSA) presents as a parkinsonian variant (autonomic failure and parkinsomism; more common in Caucasians) or as a cerebellar variant (autonomic failure and cerebellar symptoms; more common in Asians) (Klockgether, 2010).
- Sporadic adult-onset ataxia of unknown etiology (SAOA) presents with a cerebellar syndrome and reduced vibration sense.
(1) Acquired.
- Alcoholic cerebellar degeneration (including B1 deficiency) occurs in 11-27% of alcoholics and presents with severe gait and lower limb ataxia, with relatively little effect on ocular eye movement, speech, or upper limbs; MRI shows vermal cerebellar atrophy (Klockgether, 2010).
- Toxins such as phenytoin, amiodarone, chemotherapy, lithium, and heavy metals are also possible etiologies.
- Paraneoplastic cerebellar degeneration may result from almost any tumour or hematological malignancy and presents with a subacute ataxia over several weeks to months. Further immune-related etiologies include anti-GAD ataxia (associated with diabetes), gluten ataxia (associated with celiac disease), and autoimmune thyroiditis or SREAT.
- Vitamin B12 and E deficiencies often present with a neuropathy as well.
- Superficial siderosis from repeated subarachnoid bleeds in the setting of tumours or vascular abnormalitie; MRI brain shows a linear hypointensity on the surface of the brain or spinal cord (Klockgether, 2010).
- Chronic infections such as neurosyphilis, Whipple's, HIV, and CJD are possible etiologies.
(2) Hereditary.
- Friedreich's ataxia results in a sensory ataxia; MRI shows atrophy of the cervical spine.
- Spinocerebellar ataxias (SCAs) are autosomal dominant ataxias produce a cerebellar syndrome and are often associated with additional findings.
- Fragile X-associated tremor/ataxia syndrome (FXTAS) produces an ataxia with a prominent tremor, often with cognitive decline, parkinsonism, neuropathy, and autonomic failure.
- Mitochondrial disorders such as MELAS, MERFF, and NARP are possible etiologies.
(3) Sporadic degenerative.
- Multiple systems atrophy (MSA) presents as a parkinsonian variant (autonomic failure and parkinsomism; more common in Caucasians) or as a cerebellar variant (autonomic failure and cerebellar symptoms; more common in Asians) (Klockgether, 2010).
- Sporadic adult-onset ataxia of unknown etiology (SAOA) presents with a cerebellar syndrome and reduced vibration sense.
Workup
Acute Confusion Workup
(1) Do right away:
CPR.
Draw blood for FBC, electrolytes, calcium, Cr, LFTs, CRP, coags, glucose, ammonia, blood cultures.
Give thiamine 100 mg iv, dextrose 25 g iv and consider naloxone 0.4 mg (up to 1.2 mg) iv, flumazenil 0.2 mg (every minute up to 5 mg) iv.
ABG.
(2) Consider right away:
In an adult, give ceftriaxone 2 g iv, vancomycin 15 mg/kg iv, ampicillin 2 g iv and/or acyclovir 10 mg/kg iv.
Lumbar puncture (Gram stain, AFB stain, cytology, cultures, cryptococcus, VDRL, HSV-1/HSV-2/VZV PCR, glutamine).
CT/A head
Alleviate the acute confusion with dexmedetomidine 0.2-0.7 ug/kg/hour iv or haloperidol 1-5 mg 8-hourly iv or midazolam/lorazepam 1-5 mg iv bolus followed by an infusion rate of 1-5 mg per hour or quetiapine 25-200 mg o 12-hourly.
After the intravenous infusion, get a nurse special or use physical restraints, but only if necessary.
(3) Later consider:
MRI brain with gadolinium, EEG, CXR.
TSH, B1/12/E, HIV, VDRL, ANA, ENA, RF, ACE, paraneoplastic/anti-VGKC/anti-NMDA/anti-GAD antibodies, ceruloplasmin, ACTH stimulation test.
Urinalysis, urine cultures, urine porphyrins/porphobilinogen.
Lumbar puncture (OCBs, ACE, anti-NMDA antibodies, serology and PCR for CMV/EBV/HSV, VZV/JCV/enterovirus, serology for toxoplasma and mycoplasma, cryptococcus, 14-3-3 tau).
Acute Vertigo Workup
Check nystagmus (direction changing), head thrust (look for a corrective saccade), and walking (unable to walk without support).
CT/A head and neck.
FBC, electrolytes, Cr, LFTs, coags, CRP, B1, TSH, GQ1b, ceruloplasmin.
Sporadic Adult Ataxia Workup
Nerve conduction studies and EMG.
MRI brain with gadolinium (exclude superficial siderosis).
FBC, electrolytes, Cr, LFTs, CRP, HIV, VDRL, TSH, ceruloplasmin, B1/B12/E, paraneoplastic/anti-GAD antibodies, lactate.
Lumbar puncture (xanthrochromia, Gram stain, AFB stain, cytology, VDRL, Whipple's PCR, 14-3-3 tau).
Gene testing for SCA/Friedreich's/FXTAS.
(1) Do right away:
CPR.
Draw blood for FBC, electrolytes, calcium, Cr, LFTs, CRP, coags, glucose, ammonia, blood cultures.
Give thiamine 100 mg iv, dextrose 25 g iv and consider naloxone 0.4 mg (up to 1.2 mg) iv, flumazenil 0.2 mg (every minute up to 5 mg) iv.
ABG.
(2) Consider right away:
In an adult, give ceftriaxone 2 g iv, vancomycin 15 mg/kg iv, ampicillin 2 g iv and/or acyclovir 10 mg/kg iv.
Lumbar puncture (Gram stain, AFB stain, cytology, cultures, cryptococcus, VDRL, HSV-1/HSV-2/VZV PCR, glutamine).
CT/A head
Alleviate the acute confusion with dexmedetomidine 0.2-0.7 ug/kg/hour iv or haloperidol 1-5 mg 8-hourly iv or midazolam/lorazepam 1-5 mg iv bolus followed by an infusion rate of 1-5 mg per hour or quetiapine 25-200 mg o 12-hourly.
After the intravenous infusion, get a nurse special or use physical restraints, but only if necessary.
(3) Later consider:
MRI brain with gadolinium, EEG, CXR.
TSH, B1/12/E, HIV, VDRL, ANA, ENA, RF, ACE, paraneoplastic/anti-VGKC/anti-NMDA/anti-GAD antibodies, ceruloplasmin, ACTH stimulation test.
Urinalysis, urine cultures, urine porphyrins/porphobilinogen.
Lumbar puncture (OCBs, ACE, anti-NMDA antibodies, serology and PCR for CMV/EBV/HSV, VZV/JCV/enterovirus, serology for toxoplasma and mycoplasma, cryptococcus, 14-3-3 tau).
Acute Vertigo Workup
Check nystagmus (direction changing), head thrust (look for a corrective saccade), and walking (unable to walk without support).
CT/A head and neck.
FBC, electrolytes, Cr, LFTs, coags, CRP, B1, TSH, GQ1b, ceruloplasmin.
Sporadic Adult Ataxia Workup
Nerve conduction studies and EMG.
MRI brain with gadolinium (exclude superficial siderosis).
FBC, electrolytes, Cr, LFTs, CRP, HIV, VDRL, TSH, ceruloplasmin, B1/B12/E, paraneoplastic/anti-GAD antibodies, lactate.
Lumbar puncture (xanthrochromia, Gram stain, AFB stain, cytology, VDRL, Whipple's PCR, 14-3-3 tau).
Gene testing for SCA/Friedreich's/FXTAS.
Treatment Of Encephalitis, Prion Disease, Vertigo, And Sporadic Adult Ataxia
Treatable conditions should be aggressively managed.
(1) Viral encephalitis.
- In HSV and HZV encephalitis, treatment is most effective when started early. Commence acyclovir 10mg/kg iv every eight hours for 14-21 days (Tyler, 2004). The same treatment can also be used in cases of Mollaret's meningitis.
- The other viral encephalitides are not treatable beyond supportive care.
(2) Autoimmune encephalitis.
- In limbic encephalitis, treatment varies with the etiology. In patients with paraneoplastic antibodies, remove the tumour. It is also reasonable to commence methylprednisolone 1 g iv daily for five days. In patients with anti-VGKC antibodies, commence steroid therapy with methylprednisolone 1 g iv daily for five days early on (Irani, 2011). In some cases it is also reasonable to add IVIg 2 g/kg over five days.
- In anti-NMDA encephalitis, remove the teratoma if present and give methylprednisolone 1 g iv daily for five days combined with IVIg 2 g/kg over five days. If there is no response to treatment, commence rituximab 375 mg per square meter every week for four weeks; in adults, add cyclophosphamide 750 mg per square meter to the first dose and continue every month until there is a substantial clinical recovery, usually accompanied by reduced serum and cerebrospinal fluid anti-NMDA antibody concentrations (Dalmau et al, 2011). Anti-epileptic drugs should be given until there is substantial improvement, then stopped. If no teratoma was detected, continue immunosuppression with azathioprine 1-1.5 mg/kg bd or mycophenolate 1 g bd for at least one year after cessation of the initial immunotherapies (Dalmau et al, 2011).
(3) Prion diseases.
- Treatment is supportive.
- In familial CJD, genetic testing can be done if requested by other family members.
(4) Vertigo.
- For benign positional vertigo attempt the Epley's maneuver; if that fails then try prochlorperazine 10 mg o/im/iv 8-hourly or 25 mg pr bd, promethazine 25-50 mg o/im/pr 4-6 hourly or 25 mg iv 4-6 hourly, dimenhydrinate 50 mg o/im 4-6 hourly, and/or diazepam 5-10 mg o/im/iv 4-6 hourly. Steroids are of no value.
- For Meniere's attacks use the same; between episodes use low-salt diet and hydrochlorothiazide 50 mg o daily plus triamterene 25 mg oral daily.
- For acute peripheral vestibulopathy use the same plus prednisolone 20 mg bd for 10-14 days; consider prednisolone 500 mg iv if severe.
(5) Sporadic adult ataxia.
- These are often challenging.
(a) Acquired.
- Alcoholic cerebellar degeneration (including B1 deficiency) is treated with abstinence and B1 replacement.
- Toxins are treated with removal and/or chelation.
- Paraneoplastic cerebellar degeneration is treated by removing any associated tumour whereas anti-GAD ataxia, gluten ataxia, and SREAT can be managed with immunotherapy.
- Vitamin B12 and E deficiencies are treated with replacement.
- Superficial siderosis is treated by removing the bleeding source.
- Chronic infections are treated as per usual.
(b) Hereditary.
- Friedreich's ataxia is treated with idebenone.
- Spinocerebellar ataxias (SCAs) are treated supportively.
- Fragile X-associated tremor/ataxia syndrome (FXTAS) is treated supportively.
- Mitochondrial disorders are treated supportively.
(c) Sporadic degenerative.
- Multiple systems atrophy (MSA) can be treated with high-dose levodopa which usually fails; otherwise treatment is supportive.
- Sporadic adult-onset ataxia of unknown etiology (SAOA) is treated supportively.
(1) Viral encephalitis.
- In HSV and HZV encephalitis, treatment is most effective when started early. Commence acyclovir 10mg/kg iv every eight hours for 14-21 days (Tyler, 2004). The same treatment can also be used in cases of Mollaret's meningitis.
- The other viral encephalitides are not treatable beyond supportive care.
(2) Autoimmune encephalitis.
- In limbic encephalitis, treatment varies with the etiology. In patients with paraneoplastic antibodies, remove the tumour. It is also reasonable to commence methylprednisolone 1 g iv daily for five days. In patients with anti-VGKC antibodies, commence steroid therapy with methylprednisolone 1 g iv daily for five days early on (Irani, 2011). In some cases it is also reasonable to add IVIg 2 g/kg over five days.
- In anti-NMDA encephalitis, remove the teratoma if present and give methylprednisolone 1 g iv daily for five days combined with IVIg 2 g/kg over five days. If there is no response to treatment, commence rituximab 375 mg per square meter every week for four weeks; in adults, add cyclophosphamide 750 mg per square meter to the first dose and continue every month until there is a substantial clinical recovery, usually accompanied by reduced serum and cerebrospinal fluid anti-NMDA antibody concentrations (Dalmau et al, 2011). Anti-epileptic drugs should be given until there is substantial improvement, then stopped. If no teratoma was detected, continue immunosuppression with azathioprine 1-1.5 mg/kg bd or mycophenolate 1 g bd for at least one year after cessation of the initial immunotherapies (Dalmau et al, 2011).
(3) Prion diseases.
- Treatment is supportive.
- In familial CJD, genetic testing can be done if requested by other family members.
(4) Vertigo.
- For benign positional vertigo attempt the Epley's maneuver; if that fails then try prochlorperazine 10 mg o/im/iv 8-hourly or 25 mg pr bd, promethazine 25-50 mg o/im/pr 4-6 hourly or 25 mg iv 4-6 hourly, dimenhydrinate 50 mg o/im 4-6 hourly, and/or diazepam 5-10 mg o/im/iv 4-6 hourly. Steroids are of no value.
- For Meniere's attacks use the same; between episodes use low-salt diet and hydrochlorothiazide 50 mg o daily plus triamterene 25 mg oral daily.
- For acute peripheral vestibulopathy use the same plus prednisolone 20 mg bd for 10-14 days; consider prednisolone 500 mg iv if severe.
(5) Sporadic adult ataxia.
- These are often challenging.
(a) Acquired.
- Alcoholic cerebellar degeneration (including B1 deficiency) is treated with abstinence and B1 replacement.
- Toxins are treated with removal and/or chelation.
- Paraneoplastic cerebellar degeneration is treated by removing any associated tumour whereas anti-GAD ataxia, gluten ataxia, and SREAT can be managed with immunotherapy.
- Vitamin B12 and E deficiencies are treated with replacement.
- Superficial siderosis is treated by removing the bleeding source.
- Chronic infections are treated as per usual.
(b) Hereditary.
- Friedreich's ataxia is treated with idebenone.
- Spinocerebellar ataxias (SCAs) are treated supportively.
- Fragile X-associated tremor/ataxia syndrome (FXTAS) is treated supportively.
- Mitochondrial disorders are treated supportively.
(c) Sporadic degenerative.
- Multiple systems atrophy (MSA) can be treated with high-dose levodopa which usually fails; otherwise treatment is supportive.
- Sporadic adult-onset ataxia of unknown etiology (SAOA) is treated supportively.
Conclusion
Treatable sources of orientation and equilibrium disorders must be sought out aggressively. In the case of encephalitis, the difference can be drastic. In the case of HSV encephalitis, 65% of treated patients will have a favourable outcome (Raschilas et al, 2002); without treatment, 70% of patients will die (Tyler, 2004). In the case of anti-NMDA encephalitis, 79% of treated patients will recover completely, 15% will be left with a mild to severe deficit, and 6% will die (Titulaer et al, 2013); without treatment, these odds are much worse.
References
Anderson et al. 2004. Herpes simplex encephalitis. http://emedicine.medscape.com/article/1165183-overview.
Asztely and Kumlien. 2012. The diagnosis and treamtent of limbic encephalitis. Actsa Neurologica Scandinavica 126(6), 365-375.
Bataller et al. 2006. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. Journal of Neurology, Neurosurgery, and Psychiatry 78(4), 381-385.
Corato et al. 2006. Young-onset CJD: age and disease phenotype in variant and sporadic forms. Functional Neurology 21(4), 211-215.
Dalmau et al. 2011. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurology 10(1), 63-74.
Gable et al. 2009. Anti-NMDA receptor encephalitis: Report of ten cases and comparison with viral encephalitis. European Journal of Clinical Microbiology and Infectious Diseases 28(12), 1421-1429.
Geschwind et al. 2008. Rapidly progressive dementia. Annals of Neurology 64(1), 97-108.
Glaser et al. 2006. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clinical Infectious Diseases 43, 1565-1577.
Klockgether. 2010. Spodadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurology 9(1), 94-104.
Irani. 2011. Autoimmune encephalitis - new awareness, challenging questions. Discovery Medicine 11(60), 449-458.
Raschilas et al. 2002. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clinical Infectious Diseases 35(3), 254-260.
Skoldenberg. 1996. Herpes simplex encephalitis. Scandinavian Journal of Infectious Diseases 100, 8-13.
Solomon et al. 2007. Viral encephalitis: a clinician's guide. Practical Neurology 7, 288-305.
Steiner. 2011. Herpes simplex virus encephalitis: new infection or reactivation? Current Opinions in Neurology 24(3), 268-274.
Titulaer et al. 2013. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. The Lancet Neurology 12(2), 157-165.
Tyler. 2004. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes Supp 2, 57A-64A.
Vincent et al. 2004. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 127, 701-712.
Weiser et al. 2006. EEG in Creutzfeldt-Jakob disease. Clinical Neurophysiology117(5), 935-951.
Anderson et al. 2004. Herpes simplex encephalitis. http://emedicine.medscape.com/article/1165183-overview.
Asztely and Kumlien. 2012. The diagnosis and treamtent of limbic encephalitis. Actsa Neurologica Scandinavica 126(6), 365-375.
Bataller et al. 2006. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. Journal of Neurology, Neurosurgery, and Psychiatry 78(4), 381-385.
Corato et al. 2006. Young-onset CJD: age and disease phenotype in variant and sporadic forms. Functional Neurology 21(4), 211-215.
Dalmau et al. 2011. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurology 10(1), 63-74.
Gable et al. 2009. Anti-NMDA receptor encephalitis: Report of ten cases and comparison with viral encephalitis. European Journal of Clinical Microbiology and Infectious Diseases 28(12), 1421-1429.
Geschwind et al. 2008. Rapidly progressive dementia. Annals of Neurology 64(1), 97-108.
Glaser et al. 2006. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clinical Infectious Diseases 43, 1565-1577.
Klockgether. 2010. Spodadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurology 9(1), 94-104.
Irani. 2011. Autoimmune encephalitis - new awareness, challenging questions. Discovery Medicine 11(60), 449-458.
Raschilas et al. 2002. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clinical Infectious Diseases 35(3), 254-260.
Skoldenberg. 1996. Herpes simplex encephalitis. Scandinavian Journal of Infectious Diseases 100, 8-13.
Solomon et al. 2007. Viral encephalitis: a clinician's guide. Practical Neurology 7, 288-305.
Steiner. 2011. Herpes simplex virus encephalitis: new infection or reactivation? Current Opinions in Neurology 24(3), 268-274.
Titulaer et al. 2013. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. The Lancet Neurology 12(2), 157-165.
Tyler. 2004. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes Supp 2, 57A-64A.
Vincent et al. 2004. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 127, 701-712.
Weiser et al. 2006. EEG in Creutzfeldt-Jakob disease. Clinical Neurophysiology117(5), 935-951.
